
13.11.2024

PKN1 – vielversprechendes Target für die Therapie des akuten Schlaganfalls
In einer kürzlich im Fachjournal Metabolism veröffentlichten Forschungsarbeit beschreibt ein Team um die Neurobiologinnen Stephanie zur Nedden und Gabriele Baier-Bitterlich die relevante Rolle des Enzyms Proteinkinase N1 (PKN1) im zerebralen Energiestoffwechsel. Die Forscher:innen konnten belegen, dass ein Mangel an PKN1 in In-vivo- und In-vitro-Schlaganfallmodellen eine hohe Schutzwirkung zeigt – das macht PKN1 zu einem vielversprechenden Ziel für die Therapie des akuten Schlaganfalls.
Im Rahmen der Erforschung neuroprotektiver Mechanismen im Gehirn konnte am Institut für Neurobiochemie bereits nachgewiesen werden, dass Protein Kinase N1 (PKN1) durch die Regulierung des AKT/PKB Kinase-Signalwegs eine essentielle Rolle in der Gehirnentwicklung sowie bei der Neuroprotektion von Neuronenkulturen unter Sauerstoffmangel spielt. „Bei Mäusen ist das Expressions-Level von PKN1 im Gehirn kurz nach der Geburt extrem hoch. Trotzdem der PKN1-Spiegel im Laufe der Gehirnentwicklung abnimmt, ist das Enzym auch im erwachsenen Gehirn noch stark exprimiert; dort ist seine funktionelle Rolle allerdings nur wenig erforscht“, beschreibt Stephanie zur Nedden – seit 1. Oktober am Institut für Physiologie (Direktorin: Michaela Kress) tätig – die Ausgangslage der rezenten Forschungsarbeiten, die noch am Institut für Neurobiochemie (Direktorin: Christine Bandtlow) entstanden sind
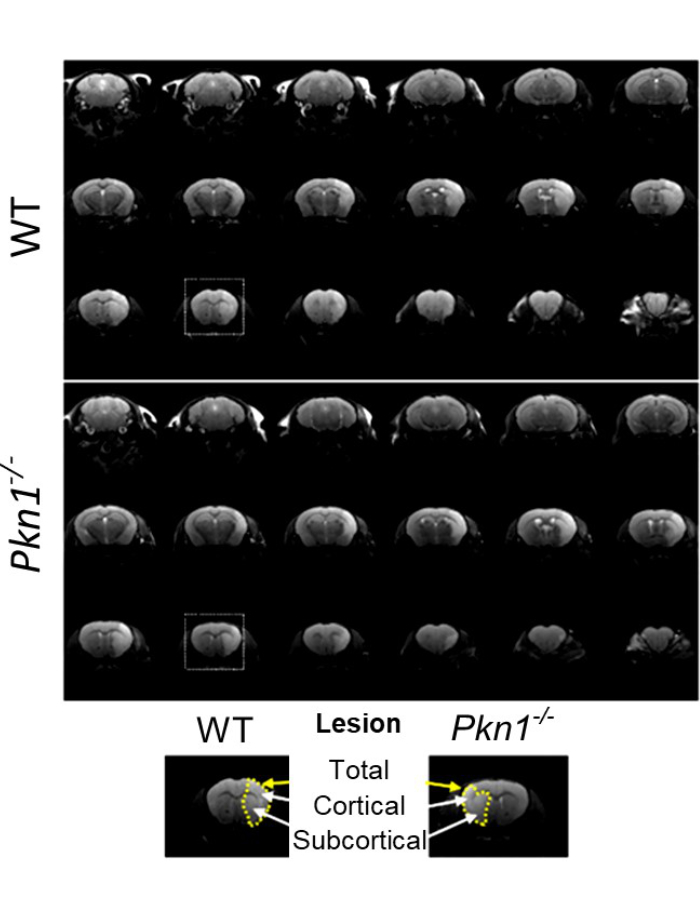
BU: MRT-Aufnahmen nach einem induzierten Schlaganfall zeigen, dass das Fehlen von PKN1 mit einer Verringerung des Läsionsvolumens einhergeht. Graphik aus der Publikation
Auf den Energiestoffwechsel kamen die Forscher:innen eigentlich über den ischämischen Schlaganfall und die Frage ob die PKN1/AKT-Interaktion auch für neuroprotektive Mechanismen relevant sein könnte. Obwohl ein Mangel an PKN1 in in vitro- und in vivo-Schlaganfallmodellen einen stark protektiven Phänotyp zeigte, ließ sich dieser jedoch nicht auf eine direkte Verbindung zum PKN1/AKT-Signalweg zurückführen.
Sie konzentrierten sich daher auf die Rolle von PKN1 im Energiestoffwechsel, insbesondere auf den Glykolyse-Stoffwechselweg und den im Mitochondrium stattfindenden Zitratzyklus. „Zum ersten Mal konnten wir eine wesentliche und einzigartige Funktion von PKN1 im zerebralen Energiestoffwechsel umfassend nachweisen, nämlich die Regulierung der Glykolyse und der mitochondriale Pyruvat-abhängige Atmung“, erläutert zur Nedden. PKN1 beeinflusst die Glykolyse direkt durch die Inhibierung von Schlüsselenzymen wie der Phosphofruktokinase. In einer weiteren Studie, veröffentlicht in Molecular Metabolism, konnte zudem gezeigt werden, dass PKN1 die Produktion von dem im Gehirn angereicherten Metabolit Glucose-1,6-bisphosphat hemmt, der eine zentrale Rolle bei der Regulation der Pyruvataufnahme in den Mitochondrien spielt.
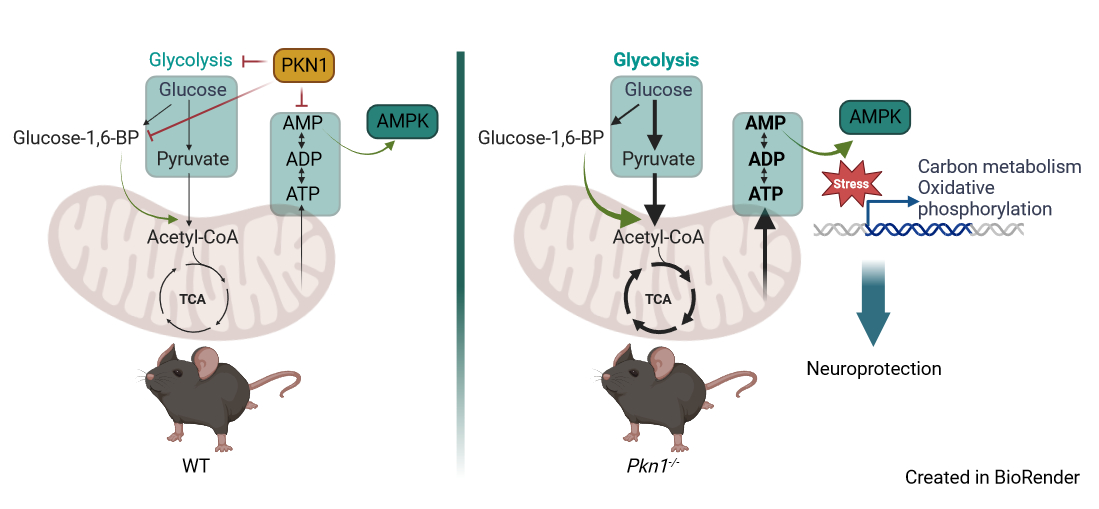
BU: Im Gehirn spielt PKN1 eine zentrale Rolle bei der Regulierung des Zuckerstoffwechsels (Glykolyse) sowie beim Einbau des Glykolyse-Produkts Pyruvat in den Zitratzyklus (TCA-Zyklus). Fehlt PKN1, kommt es zu einer Hochregulierung des Energiestoffwechsels und einer verstärkten Aktivierung des Energiesensors AMPK unter energetischem Stress, was neuroprotektive Effekte zur Folge hat. Der graphische Abstract wurde von der Originalpublikation adaptiert.
Aus diesen Gründen führte ein Mangel an PKN1 zu einer Erhöhung des zerebralen Energiestoffwechsels und damit einhergehend einer Erhöhung der Energiewährung ATP (Adenosintriphosphat). Während eines induzierten Schlaganfalls in in vitro Modellen, durch Sauerstoff-Glukose-Entzug, entstanden dadurch vermehrt ATP-Abbauprodukte, die wiederum verschiedene zelleigene Schutzmechanismen aktivierten. Ein Mangel an PKN1 führte insbesondere zu einer stark erhöhten Aktivität der AMP-aktivierten Proteinkinase (AMPK), die wesentlich zum protektiven Phänotyp in Schlaganfallmodellen beitrug.
Die Arbeiten involvierten ein großes Team am Campus die mit molekularbiologischen Methoden, RNA Sequenzierung, Analyse der Zellatmung und Untersuchungen von Energiemetaboliten bzw. Verstoffwechselung von Zuckermolekülen zur Arbeit beigetragen haben. Insbesondere waren die Core Facility für Metabolomics I (Leitung: Univ.-Prof. Dr. Herbert Oberacher), die Protein Core Facility (Leitung: Bettina Sarg) sowie die MultiOmics Sequencing Core Facility (Leitung: Anne-Margarethe Krogsdam Christensen) der Medizinischen Universität Innsbruck grundlegend an der Studie beteiligt.
Die Forscher:innen zeigen sichsehr optimistisch was die zukünftigen Möglichkeiten betrifft, PKN1 als Zielmolekül für die Entwicklung von neuen Therapieansätzen einzusetzen. „Ob sich PKN1 auch im Tiermodell hemmen lässt und als neuer protektiver Ansatz in Richtung Translation weiterverfolgt werden kann, müssen aber weitere Untersuchungen zeigen. Für die Krebstherapie ist die Entwicklung von PKN1-Inhibitoren bereits ein Thema, diese Ansätze müssten nun für den Einsatz beim Schlaganfall adaptiert werden“, schließt zur Nedden.
BU Team: v.l.: Herbert Oberacher (Core Facility für Metabolomics I), Bettina Sarg (Protein Core Facility), Klaus Faserl (Protein Core Facility), Gabriele Baier-Bitterlich, Stephanie zur Nedden, Louisa Künkel, Luisa Lang, Dido Weber und Anne Krogsdam (Core Facility für MultiOmics Sequencing).
(13.11.2024, Text: D. Heidegger, Bilder: S.zur Nedden)
Links:
Protein kinase N1 deficiency results in upregulation of cerebral energy metabolism and is highly protective in in vivo and in vitro stroke models. Stephanie zur Nedden et al., Metabolism, Volume 161.
https://doi.org/10.1016/j.metabol.2024.156039
Glucose-1,6-bisphosphate: A new gatekeeper of cerebral mitochondrial pyruvate uptake. Motahareh Solina Safari et al., Molecular Metabolism, Volume 88
https://doi.org/10.1016/j.molmet.2024.102018